Distressing Dyspnea in a Patient with Lupus
Aaron Miller, MD; Pulmonary/Critical Care Fellow, Saint Louis University School of Medicine
John Mwangi, MD; Assistant Professor, Division of Pulmonary, Critical Care and Sleep Medicine, Director of Pulmonary Hypertension Program, Saint Louis University School of Medicine
Intro:
A 59 year old female with history of systemic lupus erythematosus (SLE) presented to pulmonary clinic with worsening dyspnea on exertion. Echocardiography (ECHO) was obtained which showed normal systolic function with estimated ejection fraction of 60%. The estimated right ventricular systolic function (RVSP) was 48 mmHg. Right heart catheterization (RHC) was subsequently obtained. The pertinent RHC waveforms are included below. The pulmonary artery (PA) waveform is on the left and the pulmonary artery wedge pressure (PAWP) waveforms are on the right.
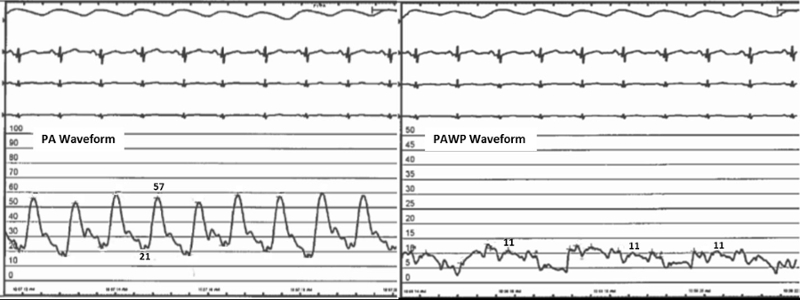
The patient’s cardiac output was measured to be 4.17 Liters/min by the thermodilution method. The mean pulmonary artery pressure is 33 mmHg.
Question:
Based on this information, what is the pulmonary vascular resistance (PVR) for this patient?
Answer:
The first step to answering this question requires the calculation of the transpulmonary gradient (TPG) using the following formula:
TPG = mean PA pressure – PAWP = 33 - 11 = 22 mmHg
The PVR can now be calculated using the following formula:
PVR = TPG/cardiac output = (22 mmHg)/(4.17 L/min) = 5.28 woods units (mmHg/L/min)
Discussion:
The patient in this case has a mean pulmonary artery pressure (mPAP) of 33 mmHg, which is diagnostic of pulmonary hypertension. The cause of this patient’s pulmonary hypertension can be further evaluated by using the PVR and PAWP. By definition, mPAP greater than 25 mmHg with low PAWP (≤ 15 mmHg) and a high PVR (>3 Wood units) is consistent with pre-capillary pulmonary hypertension.1
There are 4 main types of pulmonary hypertension that result in elevated pre-capillary pulmonary pressures. These include pulmonary arterial hypertension, pulmonary hypertension secondary to lung disease and/or hypoxemia, chronic thromboembolic pulmonary hypertension (CTEPH), and pulmonary hypertension with unclear and/or multifactorial mechanisms (Group 5 PH).1 The patient in this case underwent extensive evaluation and did not have significant chronic lung disease or chronic thromboembolic disease. In the setting of systemic lupus erythematosus, this patient’s pre-capillary pulmonary hypertension was attributed to connective tissue disease associated pulmonary arterial hypertension (CTD-PAH). According to the US REVEAL registry2, approximately 17% of patients with pulmonary hypertension secondary to connective tissue disease had a diagnosis of SLE. The 1-year and 2-year survival for pulmonary hypertension secondary to SLE is estimated to be 94% and 85% respectively.2
Management of pulmonary arterial hypertension includes an evaluation of a patient’s WHO functional class and treatment with vasodilator therapy. The patient in this case met criteria for WHO functional class 3. Vasodilator therapy was initiated using Phosphodiesterase-5 inhibitor (tadalafil), which has been shown to improve exercise tolerance and quality of life.3 Once the patient tolerated tadalafil, she was subsequently started on an endothelin receptor antagonist (Macitentan), which has been shown to delay time to clinical worsening.4
References:
-
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev Esp Cardiol (Engl Ed). 2016 Feb;69(2):177.
-
Chung L, Liu J, Parsons L, et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest. 2010 Dec;138(6):1383-94.
-
Galiè N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009 Jun 9;119(22):2894-903.
-
Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013 Aug 29;369(9):809-18.


